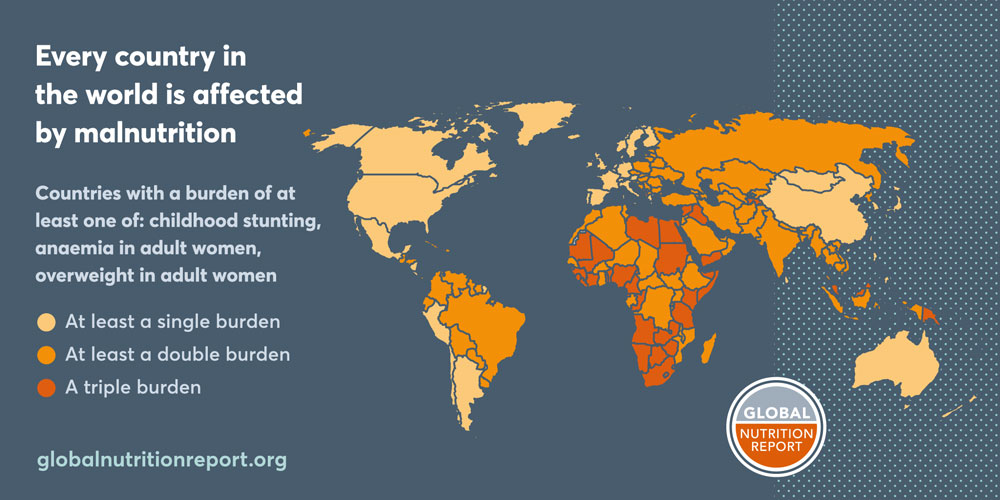
Global genomics research increases animal health metadata
Global organizations such as the World Organization for Animal Health (WOAH) and Food and Agriculture Organization (FAO) have taken the lead in compiling data related to animal disease metrics. However, as genomic data increases with the decreasing cost of sequencing, metadata related to animal health and disease can be used to supplement GBADs' existing data. There are estimates that predict that there are up to 40 billion gigabytes of genomic data generated every year and that over the next decade genomics research will generate between 2 and 40 exabytes of data (Stephens et al. 2015). The National Centre for Biotechnology Information (NCBI) is an example of a set of databases that holds information pertinent to GBADs. As more researchers are conducting genomic work on livestock and other economically important species, data on breed, location, and individual condition and disease are being recorded. In particular, NCBI's BioSamples database stores the accompanying metadata to genetic sequences uploaded to NCBI. As sequencing becomes more cost efficient, animal metadata that can be used by GBADs will accumulate. There has been a remarkable spike in the number of metadata entries concerning livestock in the past 7 years (Figure 1). These metadata accompany genomic studies carried out by not just universities, but also organizations around the world. NCBI has already been used to create databases for other researchers to use (Hu et al. 2022). The AnimalMetagenome DB (http://animalmetagenome.com) holds metagenomic data for 4 domestic species and an abundance of wild species.

Figure 1. NCBI entries in the BioSample database concerning species covered in GBADs (cattle, pig, goat, sheep, chicken, llama, equids, camel). This figure shows the pattern of number of entries since 2005.
Geographic spread of genomic data
One of GBADs' main goals is to close global data gaps concerning animal health. In order to standardize basic animal health data across countries, location and breed data are incredibly important. Classifying the global range of animals will assist in understanding the spread of diseases originating from animals. Users can input location data as a basic attribute in the BioSamples database. In particular, entries can contain the location of the submitter, sampling location, and even specific sampling coordinates. These data can reflect where certain species densely populate or which countries and locations have a lack of data. Although DNA sequencing has decreased in cost, it is still a sizable purchase; therefore, these data can also indicate whether there are inherent biases in the presence of genomic sampling across countries. Using rentrez, an R package to access NCBI (Winter 2017), we were able to extract metadata entries for genomic studies involving livestock and other economically important species. Figure 2 illustrates the geographic spread of these data, where it is shown that there are clusters of data in highly studied areas. Conversely, there are clear gaps in data across Africa and parts of Asia (Figure 2).

Figure 2. Static snapshot of an interactive figure displaying sampling locations for livestock and other species relevant to GBADs based on NCBI BioSample metadata.
Now, these are just the data that contained location information for each entry. As mentioned earlier, these data are important baseline data on global species occurrence; however, the BioSamples database also holds data more pertinent to GBADs, such as disease. We explored the mined metadata from NCBI and found that in our study species, 98 percent of entries did not contain viable disease data. By 'viable', we mean information that is disease related, as many entries had random characters or non-related information. The remaining 2 percent of data held diseases and disease agents that overlapped with WOAH's disease agent list (WOAH, personal communication of internal document). For example, bovines had 16 diseases that overlapped with the WOAH list (Figure 3).

Figure 3. Static snapshot of an interactive figure displaying sampling locations for cattle with disease information present in the NCBI entry. The diseases listed overlap with the diseases WOAH is monitoring.
Inconsistent user input leads to gaps in data
NCBI houses data for a range of organisms from bacteria to whales; however, when mining data for livestock and economically important species, close to 350,000 entries were returned. Gathering data for GBADs species (cattle, chicken, pig, sheep, goat, etc) revealed variation in the amount of data. The figure below illustrates this variation, where cattle, chicken, and pigs returned the most amount of data. The completeness of the data for these returned entries varied; however, they made up the largest proportion of GBADs-related data. Cattle entries, in particular, accounted for 27% of the total number of entries. The combination of two other species, llamas and camels, made up less than 1% of the total returned entries.
In addition to species information, NCBI also allows for users to input breed data. As breed information can be useful for understanding the prevalence and spread of disease, it is important to know which breeds form the majority of the data. Figure 4 illustrates the proportion of specific breeds for each species gathered for GBADs, where each different colour bar indicates a different breed. As shown in the plot, the majority of entries did not contain breed information. This is an important finding, as breed information should be one of the main parameters reported, especially for economically important species. This plot also shows potential gaps in the data housed in NCBI, as certain breeds are not as prevalent in the data. This can provide knowledge about where resources, time, and funding should be allocated to equalize the data across breed and species.

Figure 4. Barplot illustrating the proportion of different breeds reported for each species investigated in the NCBI BioSamples database. The pink indicates the number of entries with no breed information. All other colours indicate proportion of different breeds for each species.
Next steps in utilizing these data
Based on our first look at the available metadata on NCBI, there are a few tasks that could make the process more streamlined. Additionally, the following steps would increase metadata for secondary uses.
Standardize parameters. Standardizing parameters would improve the data cleaning process and assist in equalizing data across different institutions. Currently, the free text set up of the BioSample database makes it very difficult to compare across entries, as misspellings and extra characters can inhibit comparisons. As discussed elsewhere (Goncalves and Musen 2019), bolstering the underlying infrastructure of the NCBI BioSamples database will assist in standardizing data and make these data more accessible to researchers for secondary uses outside of genomics.
More focus on metadata. There are major gaps in data that range from smaller details such as disease prevalence to important parameters such as breed or location. As NCBI gets accessed more and more for metadata, it will be imperative that users input as much data as possible. Basic data such as species, breed, and location should be mandatory for users to input.
Preprint data uploads. Currently, there is a lag between sampling and DNA sequencing and when these data get uploaded to public databases such as NCBI. This lag can vary from a few months to years. Therefore, data that could be used in secondary projects such as this one could be released up to a few years after sampling. Therefore, I recommend uploading genomic data with its accompanying metadata to NCBI as soon as possible or when preprints are submitted.
NCBI and other genomic databases hold data that can be useful for more than just genomic projects. For GBADs, these data can provide valuable information about presence of local breeds in normally unsampled locations as well as occurrences of diseases in sampled individuals. Additionally, it provides the first look into how genomic projects may supplement GBADs' aim to gather animal health data and metrics.
References:
Goncalves R, Musen MA (2019) The variable quality of metadata about biological samples used in biomedical experiments. Scientific Data, 6, 190021.
Hu R, Yao R, Li L, et al. (2022) A database of animal metagenomes. Scientific Data, 9, 312.
Stephens ZD, Lee SY, Faghri, F, et al. (2015) Big Data: Astronomical or Genomical? PLoS Biology, 3, 1002195.
Winter, DJ (2017) rentrez: An R package for the NCBI eUtils API. The R Journal, 9, 520-526.

 Figure 1: The Roadmap to Reproducibility
Figure 1: The Roadmap to Reproducibility Figure 2: Factors that contribute to irreproducible research. Figure obtained from Baker, 2016
Figure 2: Factors that contribute to irreproducible research. Figure obtained from Baker, 2016